
深入解析:三类医疗器械公司注册要求及其相关法规遵循指南
一、了解三类医疗器械的定义与范围
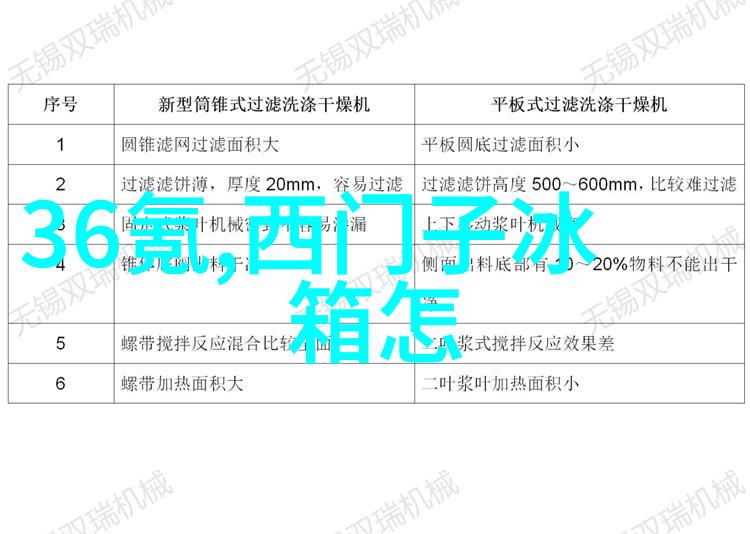
三类医疗器械通常是指那些对人体没有直接接触的设备或材料,它们可能包括一些辅助性设备或者不涉及直接人体接触的材料。这些产品由于其低风险特性,受到较为宽松的监管。

二、注册流程中的主要环节概述
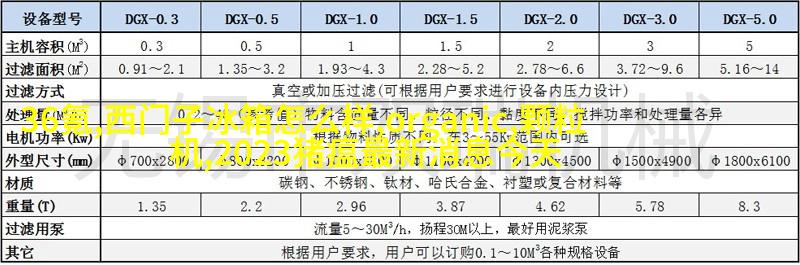
在中国,三类医疗器械的注册过程需要通过国家药品监督管理局(NMPA)的审批程序。在这个过程中,申请者需要提供详细的产品信息和测试报告,以确保产品符合安全和有效性的标准。

三、生产环境和质量控制标准
为了保证产品质量,生产环境必须符合相关法律规定。包括清洁条件良好、防尘防污等。此外,质量控制措施也非常重要,如有机合成物质检测、微生物检测等,以确保最终产品安全可靠。
四、标签和说明书编制要求
标签和说明书应当清楚地描述产品用途、使用方法以及注意事项。同时,还需明确告知用户如何正确存储和维护该种医疗器材,以避免潜在风险。

五、新兴技术在三类医疗器械上的应用
随着科技进步,一些新型材料或制造工艺被引入到三类医疗器械领域。这意味着可以开发出更先进、高效率且成本更低的人体健康支持工具,但这也增加了对这些新技术适用性的考察需求。
六、未来发展趋势与挑战分析
随着全球化市场竞争加剧,对于高质量医疗用品需求日益增长。因此,将来可能会出现更多创新型、三类医疗器械公司,并将面临更严格的国际标准检验,同时还需不断适应消费者对于便捷性与效果之间平衡需求的变化。



